
Menu:
My long term research goal is to change the way scientists analyze high dimensional biomedical data for the goal of scientific discovery. The rate of change in technology that advances our ability to collect observations about genomic data, including DNA, single cells, and tissue samples, rapidly makes analytic methods for these observations obsolete. Furthermore, the complexity of the biological phenomena we attempt to quantify and understand overwhelms current methods that oversimplify the complexity in order to scale to the data magnitude. General approaches to data analysis, including principal component analysis and linear regression, are insufficient for the intricacy of modern biomedical data; new approaches using statistical models and machine learning methods that include analysis- and technology-specific structure must be developed for many types of genomic studies.
My group builds and applies structured hierarchical models and approximate methods for the analysis of high-dimensional genomic data. Our work in developing methods for modern genomic technologies and scientific questions requires three types of innovations. First, statistical models need to be adapted to capture the complexity of the data. Second, inference algorithms for these structured models need to scale to the size of the data. Third, software infrastructure must be usable by the biomedical community. The impact of addressing these issues is that the pace of discovery and actionable results from biomedical research is accelerated, because the analytic solutions from advanced platforms are broadly available and immediately applicable. The development of these frameworks is specific to technology and analytic goals, and is not easily generalized. To this end, our work has broadly focused on innovations in two types of statistical analyses: structured regression models for hypothesis testing, and hierarchical latent variable models for dimension reduction and exploratory data analysis, as detailed below.
To this end, my work has broadly focused on innovations in two types of statistical analyses: structured regression models for hypothesis testing, and hierarchical latent variable models for dimension reduction and exploratory data analysis. Along with the development of these frameworks comes adaptations of inference methods for robust and tractable posterior inference in these models by using ideas from machine learning, and validation of the latent structure and hypothesis testing using experimental validation.
Multiple hypothesis testing frameworks for quantitative genetics
The field of quantitative genetics aims to understand the genetic basis of complex traits including human disease; in order to accomplish this aim, statistical tests must be developed to identify associations in studies with limited sample sizes between genotypes and quantitative or binary traits. These studies often include many traits and whole genomes, pushing the number of statistical tests into the trillions and requiring reformulation of the corrections for multiple hypothesis testing methods. Moreover, studies of complex traits include technical and biological confounders, such as batch effects, population structure among the samples, or variance due to age, sex, or body mass index.
Statistical tests for functional genomics.
- Expression quantitative trait loci (eQTLs) are genetic mutations that regulate gene transcription and often drive disease risk. As a postdoc, I was involved in an early eQTL paper using data from RNA-sequencing; my contribution was in the statistical methodologies for eQTL discovery in the presence of population structure [Pickrell et al., 2010].
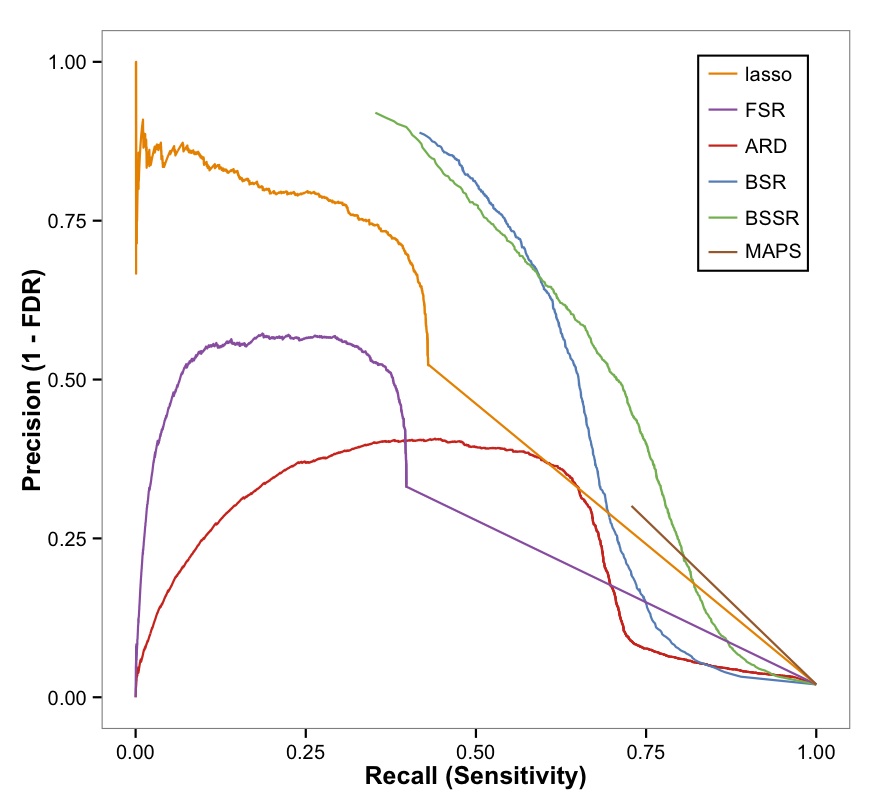
- I developed a statistical model for differential eQTLs, or eQTLs that are regulatory under one condition but not another. We found six differential eQTLs in our study. The strongest differential eQTL, which was regulatory after exposure to statins but not in their absence, was found to be protective of muscular myopathy in two separate studies of the myopathic toxicity of statins [Mangravite, Engelhardt, et al., 2013].
- With Julien Ayroles, we have developed a test for variance QTLs using a Bayesian heteroskedastic model and using iterated Laplace approximations for computation of the Bayes factor [Dumitrascu et al., 2015].
- I was involved in work to understand the genetic basis of response to d-amphetamine from high-dimensional and noisy time series survey data. In this work, we developed a sparse latent factor model to capture each subject's unique response using ten interpretable response types. With this approach, we identified a novel genetic association with the low-dosage, positive response to \emph{d}-amphetamine. Importantly, we found that our genetic association was also protective of schizophrenia and attention deficit hyperactivity disorder. As a part of the Genetics of Personality Consortium, I was involved in two meta-analysis papers across multiple GWAS that study the genetic basis of neuroticism and found a relationship with major depressive disorder risk, and the genetic basis of extraversion [Hart, Engelhardt et al., 2012] [Hart et al., 2014] [de Moor et al., 2015] [van den Berg et al., 2016]
- We developed a canonical correlation analysis with a structured prior to identify associations between multiple observation types from the same set of samples, and we used this to identify collections of genetic variants associated with collections of genes [Zhao et al., 2016].
- We developed an association test between covariates, including genotypes, and traits collected over time, such as gene expression response to perturbation, using a Gaussian process regression model. This model naturally allows the incorporation of covariates [Tonner et al., 2016].
Publications: [Engelhardt & Adams 2014]
Sparse latent factor models for recovering latent structure in genomic data
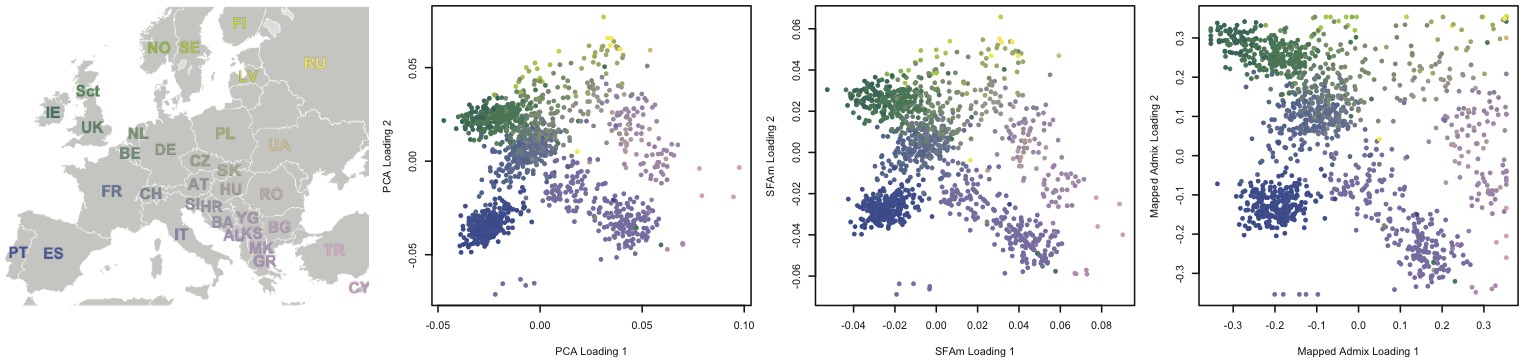
- Sparse latent factor models applied to genomic data have the ability
to recover interpretable latent linear structure. Applied to genotype
data from individuals with discrete population structure, we can
recover the underlying ancestral populations; applied to individuals
with continuous population structure, we find a recapitulation of
their geographic ancestry [Engelhardt & Stephens, 2010].
- We developed latent factor models for application to gene expression data, adapting flexible continuous sparsity-inducing priors to support an overcomplete represetation and recovering a large number of sparse latent components. We also added a two component mixture model to support recovery of non-sparse, low rank structure, which captures variance effects due to confounding such as population structure and technical effects [Gao, Brown, Engelhardt 2013].
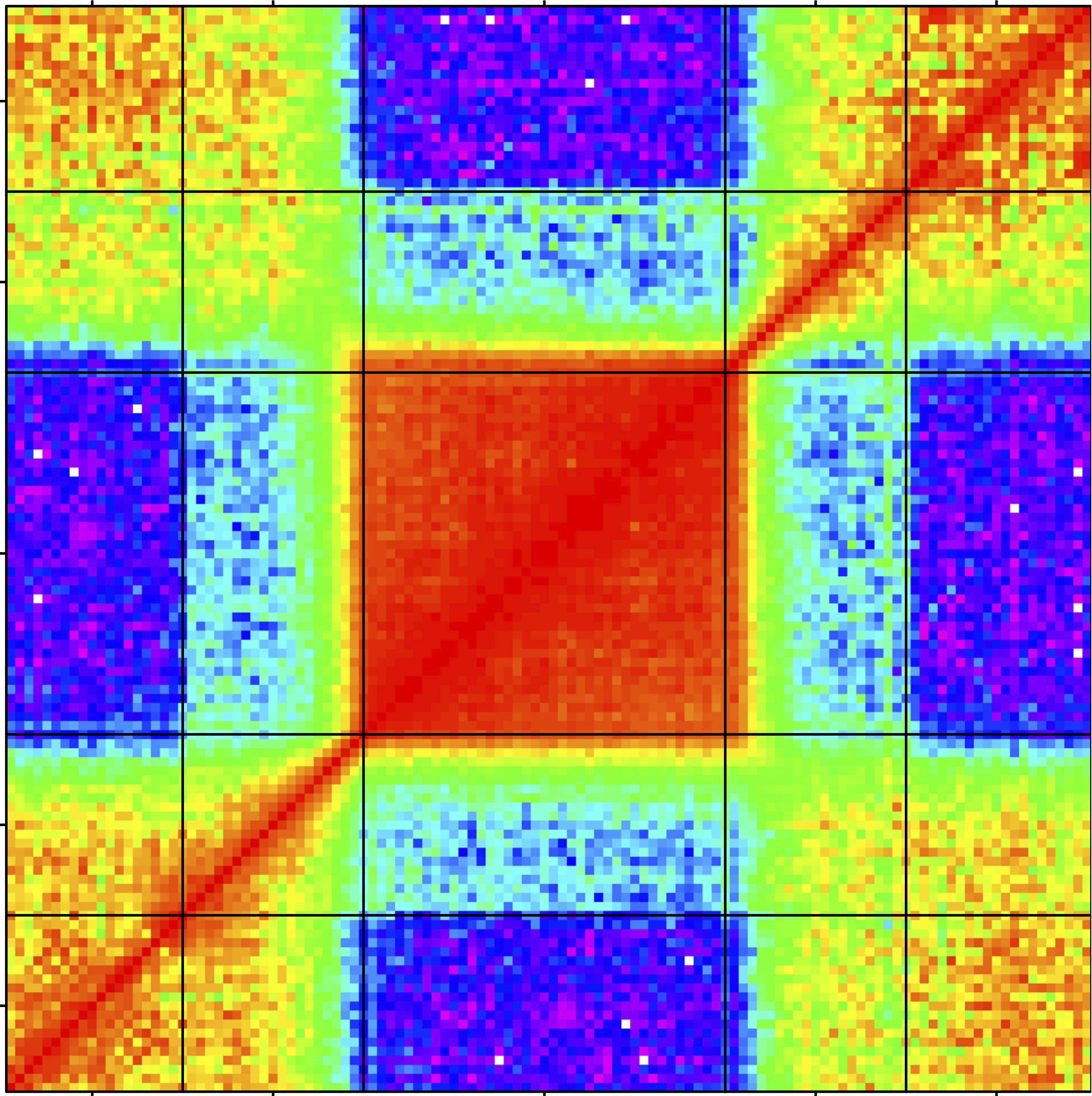
- Using this sparsity prior, we have developed
biclustering models with
sparsity on both the genes and the samples. By interpreting the latent
structure as regularized covariance matrix estimation, we build
ubiquitous, subset specific, and subset differential Gaussian
graphical models (Gaussian Markov random fields, gene co-expression
networks) [Gao et al., 2016].
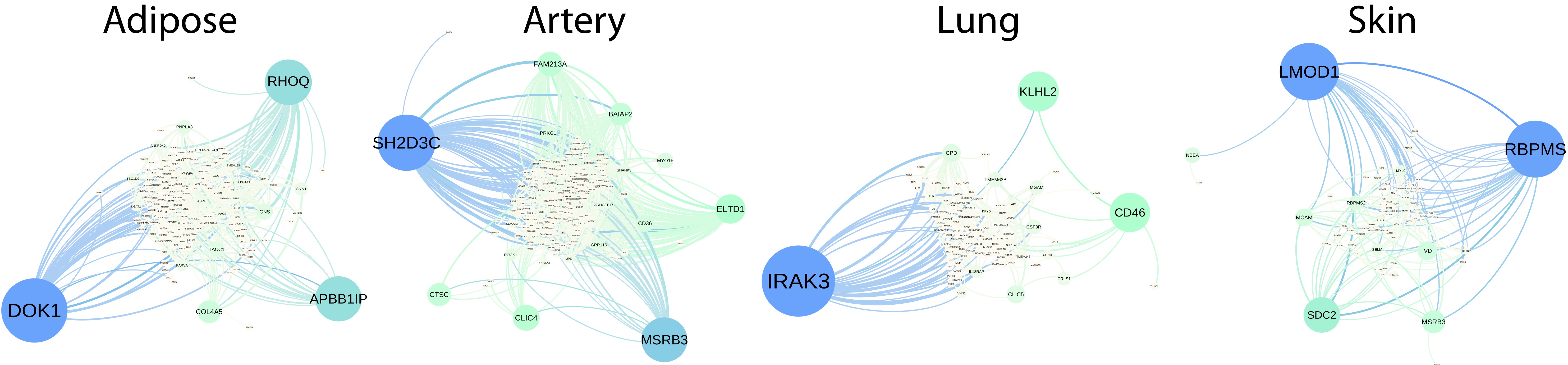
- Often these observations contain a high proportion of observations that are missing non-uniformly at random (single cell RNA-seq; Netflix ratings). We developed the hierarchical compound poisson factorization (HCPF) model to address this issue, explicitly modeling the missingness structure of the data and sharing information between the model of the observations and the missingness model [Basbug and Engelhardt, 2016]
- We extended the HCPF to allow observations across time using a gamma-Markov chain [Jerfel et al., 2017]
- Sparse latent factor models generally require a choice of the number of latent components. We developed a nonparametric latent variable model using a generalized double Pareto prior and fast inference to address this problem in a computationally tractable expandable factor analysis model [Srivastava et al., 2017]
- We are very interested in methods for model checking for latent variable models. We proposed an approach using posterior predictive checks for the admixture model of genomic data, where the discrepancy functions correspond to population genetic statistics of interest .
Studying the mechanistic underpinnings of functional SNPs
In order for SNPs associated with complex traits and disease to be medically actionable, it is essential that we understand how they work.
- We conducted large-scale replication studies across eleven studies in seven tissue types. We have overlaid these results onto regulatory element data to enable a much more profound mechanistic understanding of eQTL data by studying where eQTLs and cell type specific eQTLs are co-located with specific cis-regulatory elements [Brown et al., 2013].
- As a PI in the GTEx Consortium, my group is involved in the association mapping for trans-eQTLs and the downstream functional analyses [Jo et al., 2016].
Electronic medical record and hospital inpatient data
- With UPenn Hospitals, we are working with records from over 260,000 inpatients, including patient demographics, vital signs, lab test results, and major events. We have designed a multivariate Gaussian process regression model to smooth, impute, and predict future patient state for doctors in a hospital setting using a structured kernel [Cheng et al., 2017].
- We developed an off-policy reinforcement learning approach to identify the optimal policy to wean patients from ventilators. This is particularly difficult as we assumed the doctors' policies were suboptimal; validating our learned policy is challenging in this situation [Prasad et al., 2017]
- To ensure that statistical inference in these high-dimensional Gaussian processes was feasible, we developed Large Linear GP (LLGP), an extension of the linear model of co-regionalization to multi-output GPs that exploits the structure of the Gram matrix and naturally allows nonstationary processes [Feinberg et al., 2017].
Epigenome-wide association studies
We are currently developing methods for performing epigenome-wide scans for association of methylation status with phenotypes of interest. Current developments involve developing and applying methods for causal inference to unravel the relationships between epigenetic effects.Publications: [Zhang et al. 2015]
Protein molecular function prediction
As a graduate student with Dr. Michael Jordan, collaborating with Dr. Steven Brenner, I created a statistical methodology, SIFTER (Statistical Inference of Function Through Evolutionary Relationships), to capture how protein molecular function evolves within a phylogeny in order to accurately predict function for unannotated proteins, improving over existing methods that use pairwise sequence comparisons. We relied on the assumption that function evolves in parallel with sequence evolution, implying that phylogenetic distance is the natural measure of functional divergence. In SIFTER, molecular function evolves as a first-order Markov chain within a phylogenetic tree. Posterior probabilities are computed exactly using message-passing, with an approximate method for large or functionally diverse protein families; model parameters are estimated using generalized expectation maximization. Functional predictions are extracted from protein-specific posterior probabilities for each function. I applied SIFTER to a genome-scale fungal data set, which included families of proteins from 46 fully-sequenced fungal genomes, and SIFTER substantially outperformed state-of-the-art methods in producing correct and specific predictions.
Publications: [Engelhardt et al., 2006], [Engelhardt et al., 2007], [Engelhardt et al., 2011]