- Saccharomyces cerevisiae (brewer's yeast)
Eschericia coli
Homo sapiens (human)
Zea mays (maize, or corn)
Dictyostelium discoideum (slime mold)
As an example, if you feel that the correct order of elements were:
ORF, stop codon, promoter... your answer would be C,F,B...
| A. Ribosome Binding Site | 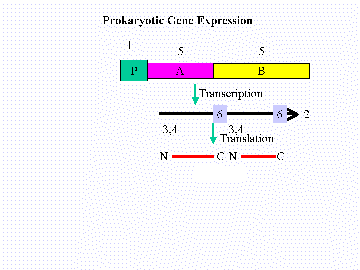 |
| B. Promoter | |
| C. cistron (ORF) | |
| D. Polycistronic mRNA | |
| E. Initiation codon | |
| F. Stop codon
|
| A. PolyA tail | 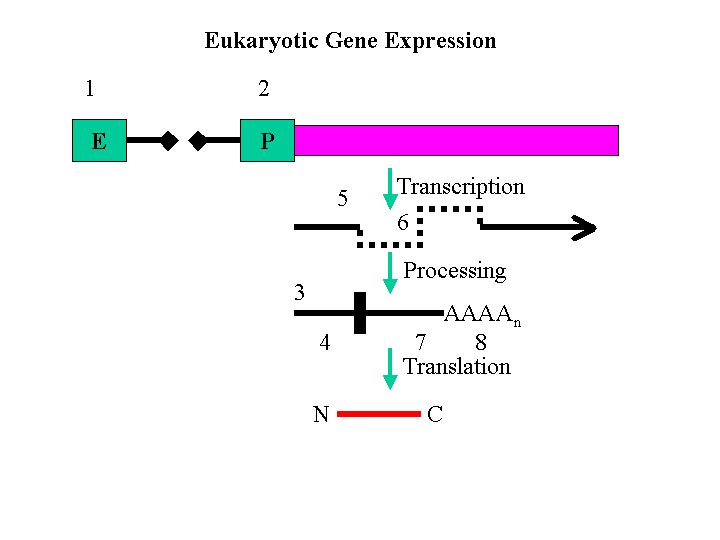 |
| B. Intron 1 | |
| C. 5'-Untranslated Region | |
| D. Initiation codon | |
| E. Stop codon | |
| F. Basal Promoter | |
| G. Enhancer | |
| H. Exon 1 |
polypeptide that is five amino acids long.
TAC ATG ATC ATT TCA CGG AAT TTC TAG CAT GTA ATG TAC TAG TAA AGT GCC TTA AAG ATC GTA CAT
Which strand of DNA is transcribed, and in which direction?
Label the 5' and 3' ends of each strand.
If an inversion occurs between the second and third triplets from the
left and right ends, respectively, and the same strand of DNA is transcribed,
how long will the resultant polypeptide be?
Assume that the original molecule is intact and that transcription
occurs on the bottom strand from left to right. Give the base sequence,
and
label the 5' and 3' ends of the anticodon that inserts the fourth amino
acid into the nascent polypeptide. What is this amino acid?
- Genome size in kilobasepairs of the human genome
Genome size in kilobasepairs of the E. coli genome
Number of amino acids in a "typical" protein.
Size, in kbp, of a "typical" human gene
Number of genes in the E.coli genome
Number of genes in the human genome.
Go to the Entrez database browser at the National Center for Biotechnology Information (NCBI). Search in the protein database for p53. I suggest you specify protein name "p53" and Organism "human" (i.e. p53 [Protein Name] AND human [Organism]). You can do this in two steps. If you just specify protein name = p53 you get p53 from the crab-eating macaque and many other interesting creatures as well. You should end up with about three documents, which you can retrieve. Find the Entrez record for human p53 with Accession number AAC12971 and display it in GenPept report format. This tells where the database submission came from, and gives information about the protein, including citations to the scientific literature. The actual sequence of amino acids is at the bottom of the report.
Q1: How many amino acids are there is the human p53 protein?
Now leave the GenPept report and go back and look at the other types of reports for this protein. The FASTA report is a very simple, machine readable format file consisting of a description line beginning with ">", in this case giving various names and AC. numbers for this protein, followed by the sequence of amino acids on one or more lines. Most bioinformatic analysis programs accept input sequences in FASTA format. The ASN.1 report is a more complex, structured machine readable information file. This format is standard at NCBI.
Now try the one nucleotide link [U94788.1]. Now you have gone from the protein world to the DNA world. You have left the protein sequence database GenPept and are in GenBank, the nucleic acid sequence database. Click on the GenBank report. This record describes the human gene that makes the p53 protein. The DNA sequence for the gene is given at the bottom. As discussed in class, human genes consist of exons and introns. These are better viewed in the graphical display. Click on the [U94788] at the top of the report. The thick light blue line represents the entire GenBank entry for this gene (the "OVERVIEW line"), and the other lines show the locations of key features in this gene. The magenta rectangles are coding exons (CDS). These are parts of the gene that are translated by the ribosome into amino acids. The thinner dark blue rectangles are the exons in the mRNA sequence made by this gene. Note how they correspond to the coding exons, except that the last one extends further (this extension is the 3' untranslated region (UTR) of the mRNA) and there is an extra exon quite a distance before the first coding exon (this is part of the 5' UTR).
Q2: How many base pairs are there in this GenBank entry for the human p53 gene? How many coding exons? How many (mRNA) exons? Where do they occur?
Click on the thick light blue OVERVIEW line at a location just above the first coding exon. This should zoom in to a region between bases 10,000 and 12,000 (about).
Q3: Does the first coding exon coincide with the second mRNA exon, or does it contain some part of the 5' UTR? What is the amino acid sequence produced by the first coding exon, and what bases of the 5' UTR are included in the corresponding mRNA exon, if any?
You may find that it helps to click again on the OVERVIEW sequence right above the first coding exon and zoom in further to answer this question! This last, most-zoomed graphic display is nice, because it shows the central dogma of molecular biology, DNA -> RNA -> protein, in action. Note also that some alleles are marked. Some people have different versions of this gene.
Go to the SWISSPROT database and find the record for the human p53 protein (P53_human) "by description". You see at least 47 references to the literature. After this, the comments field tell us, among other things, that p53 acts as a tumor suppressor, and its normal function is to stop cells from growing, or to die at the right time (apoptosis). When something goes wrong with p53, cells can grow in an uncontrolled manner, a hallmark of cancer. Scan to near the bottom of the record, and you will find a list of many mutations of the p53 gene that cause it to make a different amino acid at some position in the protein, making the person prone to getting cancer. These are usually SNPs (single nucleotide polymorphisms) that cause a substitution of one amino acid for another. Find the tumor-causing substitutions of R (arginine) at position 110.
Q4: What amino acid substitutions of the R at position 110 in the p53 protein are listed as involved in cancers? What SNPs might cause these?
To answer the last question, you will need to go back and find the three nucleic acids in p53 that form the codon that makes the R in position 110 in p53. Then you will have to look in a table of the genetic code to see what codons code for these other amino acids.
SWISSPROT gives extensive cross-references to other databases, including GenBank and the mirror site at EMBL (European Laboratory for Molecular Biology), PIR (protein Information Resource), and PDB, the Protein Data Bank, a database of three-dimensional protein structures. We'll look at this later in the assignment. For now, find the PFAM entry in the p53 SWISSPROT record and click on it. PFAM is a database of multiple alignments of related protein sequences. As discussed in class, sets of protein sequences that have evolved from a common ancestor are very useful in understanding and predicting aspects of protein structure and function. Click on "Get alignment". (The default view is fine. Select Jalview for a fancier multiple alignment viewer if you want to explore this further. Below I only refer to the default multiple alignment view.) You see an alignment of 12 protein sequences. One is P53_human; the rest are similar proteins from different organisms. Dots are inserted so that the corresponding amino acids from all twelve organisms line up in columns. Scan across, and note that some regions of the protein are more highly conserved than others.
Find the arginine at position 110 of human p53 in this alignment. It is about 1/3 of the way through. (If you clicked on one of the substitutions for this amino acids on the SWISSPROT page, then you got a context which told you that QGSYGF precedes this arginine and LGFLH follows it. This is useful in checking if you have the right arginine.)
Q5: What other amino acids occur in this position in the other organisms listed in this multiple alignment? (list them). These amino acid substitutions probably do not disrupt p53's function, since they are tolerated in these other organisms. However, the SWISSPROT file for human p53 lists 3 tumor-associated substitutions for position 110. Presumably these are disruptive. Are there amino acid properties that distinguish the (presumably) disruptive from the (presumably) non-disruptive substitutions? Which properties?
Now repeat the previous two questions, but instead use the arginine at position 248 in the human p53 protein. You'll find that substitutions of this residue also make a person prone to cancer. In this case there are even more disease-associated substitutions. This residue occurs about 2/3 of the way through the alignment. This region of the protein is highly conserved among the 12 species, and in particular, all twelve proteins have arginine in this position. One might conclude that perhaps the residue in this position of the protein must be arginine for the protein to function and for the organism to be healthy. In fact, if you only look at human p53 and it's very close orthologs (corresponding proteins in different species, presumably descended from a common ancestor protein), it seems like the whole sequence or residues MCNSSCMGGMNRRP (and perhaps more) is completely conserved. For many years people used the conserved "motif" MCNSSCMGGMNRRP that occurs in p53 at this place as a signature sequence of p53, searching for this string in proteins from other organisms to find orthologs of p53. This string produced few false positives (proteins that have this motif in them but are not orthologs of p53) and few false negatives (proteins that are orthologs of p53 but do not have this motif in them.) However, looking at the more distant members of the family in this alignment, in particular, the last sequence, which is from a squid, we see that many positions in this motif can vary. Very few individual residues in a typical protein are absolutely essential, in that no substitutions exist that preserve function. Arginine 248 in human p53 may be one of them, but some of its adjacent amino acids certainly are not. In general, distantly related orthologs cannot be found by searching for "signature" sequences like this. Either the signature is too short, in which case you get too many false positives, or the signature is too long, in which case you get too many false negatives.
In the early days, Amos Bairoch, the designer of SWISSPROT, and his collaborators put a lot of effort into developing generalized "signature" motifs that allow particular substitutions in particular places in the motif, in hopes of finding motifs that would have no false positives or false negatives for a given protein family. The motif database they produced is called PROSITE. If you click on the prosite link from the SWISSPROT p53 page you can see on the PROSITE record that they proposed the completely conserved motif MCNSSCMGGMNRRP as a signature motif for the p53 family, and they tested this pattern at the time this work was done, concluding that it found no false positives or false negatives. However, the database has grown considerably since then, as has our ability to locate likely orthologs.
Now go back to the SWISSPROT record for human p53 and find the list of PDB entries. Click on the ExPASy link for 1TSR. This gives access to information about the structure of the p53 protein from PDB. PDB is the Protein Data Bank, a repository of protein structures solved by x-ray crystallography or by NMR. Each solved structure has a 4 letter identifier. This is the PDB record for 1TSR. This particular structure is p53 bound to DNA. There is a lot to explore from here, but first note that there is a link to MMDB, NCBI's structure database. Follow this link to PubMed and on from there to retrieve the MEDLINE abstract for the paper in Science that describes the 1TSR structure. Notice that 1TSR is the structure for the core DNA-binding domain of the protein (here defined as residues 102-292) bound to a piece of DNA. p53 is a DNA binding protein that can influence another protein by binding in front of (i.e. on the 5' side) of its gene and thereby altering the way the gene for that other protein is transcribed, in this case by causing it to make more copies of the protein. In this structure, p53 is "caught in the act", so to speak.
Now go back to ExPASy PDB page for 1TSR. Click on "3D images: ribbons" to see a picture of the structure of p53. Click on "Download/Display File" and download the full PDB file with coordinates of all atoms in the 1TSR structure. To view the PDB structures, you can install the free RASMOL viewer or use the one we have installed on the ???? machines in the CS department. (You'll need to make sure you have an account on these machines) The actual RASMOL program is called ?????? machines.
The RASMOL program has on-line help. Unfortunately our installation does not seem to have it wired in. Open the file /projects/compbio/doc/rasmol/rasmol.html using your browser and you will get a RASMOL reference manual.
There are two windows when running RASMOL, a display window that contains the graphic image and a separate command window. To experiment, set the display in the display window to various modes (cartoon is a good place to start and spacefill is fun). Rotate the molecule by using the sliders on the edges of the display window.
The DNA double helix is visible in yellow, and you can see 3 separate protein chains all of similar structure. Each of these is a p53 core domain. They are chains A,B and C. Color chain B red by typing the following commands in the command window:
select *:B color redTo see particular residues, say residue 110 (the arginine at position 110) in chain B, type
select 110B color yellowThis is the residue you were looking at previously in the PRODOM multiple alignment and in the SWISSPROT file. Another good one is residue 248 in chain B. This is the conserved arginine at position 248 we explored above. Select that one and color it green. Under the "options" menu on the display window, choose "Labels". This shows the amino acid name for the residue(s) you just selected. I suggest you choose "spacefill" under the display menu as well, so you can see the whole shape of this amino acid. To zoom in to get a closer look, rotate the structure to center this residue, and type
zoom 400This magnifies 400%.
Q6: Looking at this arginine at position 248, where is this arginine in the structure relative to the DNA sequence? This residue was in the critical, totally conserved region of p53. Why do you think it is so important?